
地址:镇江市丹徒新城恒园路67号
手机:13805284038
E-mail:zjgl@zj-boiler.com
客服热线:13861390288
版权所有 - 江苏昆仲机械有限公司 | Copyright - 2018 All Rights Reserved

新闻动态
NEWS
公司新闻
行业新闻
ASPEN物性参数计算中子模型的调整方法
ASPEN物性参数计算中子模型的调整方法
二甲基镉,是一种无色剧毒的液体,在空气中形成烟雾。二甲基镉被许多化学家认为是人类已知的毒性最强的化学物质,因为它不止剧毒,还是一种高爆炸性、高致癌性物质。
二甲基镉主要用作烷基化等有机合成及聚合的催化剂,也用于金属有机化学沉积法(mocvd)制造半导体异质结材料,。
目前国内生产二甲基镉的厂家主要有:大连光明气体研究所和南京大学一试剂公司,由于半导体工业的需要,南大光电似乎也在生产该物质。
根据文献查询发现,二甲基镉的物性参数极少。但由于高纯度半导体级二甲基镉的纯化过程中,需要大量的物性参数,如蒸发焓、液体粘度、表面张力,以及计算过程中由于内插或者外推过程中所需的不同温度压力下的上述参数所需的临界压力、临界体积、偏心因子等数据。缺乏这些数据对于设计人员是个极大的挑战。
ASPEN不仅具有工程设计计算功能,而且还具有物性参数估算功能。ASPEN通过基团贡献法或各种经验公式对各种物性参数进行估算。这些理论的或经验的公式包含在物性方程中,一个物性方程包含各种各样的模型方程,如预测液体体积、蒸发焓、表面张力、粘度的相应方程。而这些子模型方程中有许多种文献报道过的比较可靠的预测公式可供选择,默认情况下,系统给出的选择是目前比较常用的普适性方程。而这个普适性方程是否适合某一物质则需要使用者慎重选择。
下面就作者在设计二甲基镉(DIM)-四氢呋喃(THF)的精馏分离中遇到的问题之一—液体密度问题进行分析,抛砖引玉,从中得到一些启发。
1 根据系统默认子模型计算液体密度
已知的DIM物性极少,已知情况如下:
表1 二甲基镉(DIM)已知的物性参数
|
项目 |
值 |
项目 |
值 |
|
分子量(MW) |
142.481 |
正常沸点(TB) |
105.5℃ |
|
密度(ρ) |
1986(17℃) |
三相点(TPT) |
-2.668 |
|
融化热(HFUS) |
1.87174kcal/mol |
|
|
组分中只输入DIM的组分名,选定物性方程为WILSON方程,利用系统的analysis功能分析不同温度下液体密度,如图1所示。
点击运行后,很遗憾,因为缺乏某些参数,分析无法运行,如图2所示。根据控制面板错误信息提示,系统缺乏Pc、Zc导致无法运行。同时,提示显示预测液体密度模型是VL0RKT。

图1 analysis分析功能对液体密度分析

图2 控制面板出现的错误信息提示
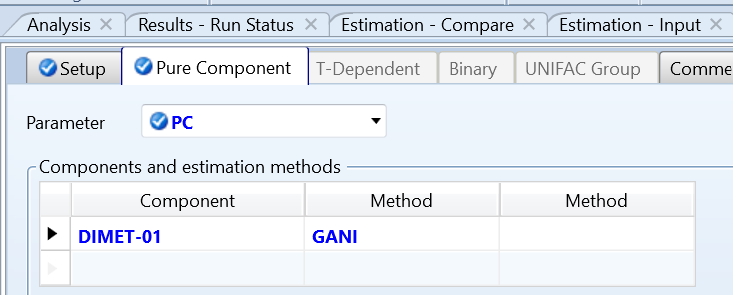
图3 在“ESTIMATION”估算系统中对Pc、Zc进行估算
运行估算系统后,在Methods>Parameters>Pure components目录下自动出现一个PCES-1项,如图4所示。
 |
图4 PCES-1显示的估算项目
图中可以发现先前设置的估算项目,Pc、Vc、Zc三项及其估算值,其中Vc是估算Zc时需要用到的参数。
再次运行analysis分析功能,得到其结果如图5所示。
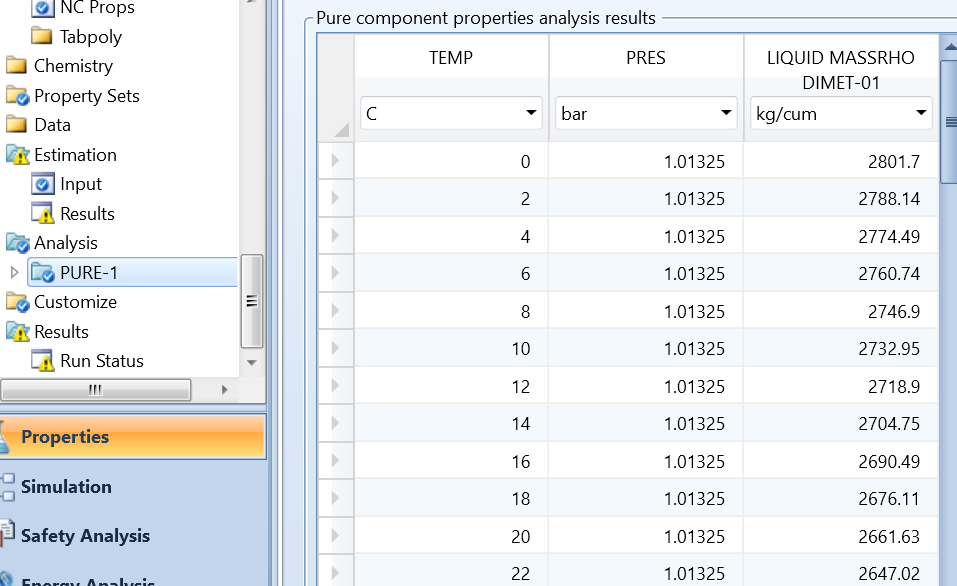
图5 analysis对密度的分析结果
由图5可以,发现17℃时,预测密度约为2680kg/m3,与实际相差很大。
在此情况下,我们需要修改wilson方程模型中系统默认的密度预测模型VL0RKT,如图6所示。VL0RKT模型采用Rackett、DIPPR、NIST、IK-CAPE、PPDS五个方程之一计算物质的液体体积,其中系统默认方程为Rackett,因此,前述的系统错误提示中显示需要Pc、Zc的值。
2 models 下调整预测液体密度公式
如图7所示,在Methods>Parameters>Pure components>THRSWT-1,中,第二项是计算液体密度的公式选项,0是Rackett的代号,100-116是DIPPR不同公式的选项,选择105运行分析,结果错误。
控制面板信息如图8所示。图中显示缺乏DNLDIP,这是液体摩尔密度系数,而我们需要计算的是液体密度,显示待求变量和输入变量是一回事,说明这个公式此时对于求解液体密度无能为力。
我们再将图7中代码选为302,希望用PPDS Campbell-Thodos计算。其需要的输入变量如表2所示。需要的摩尔体积可用临界摩尔体积代替。
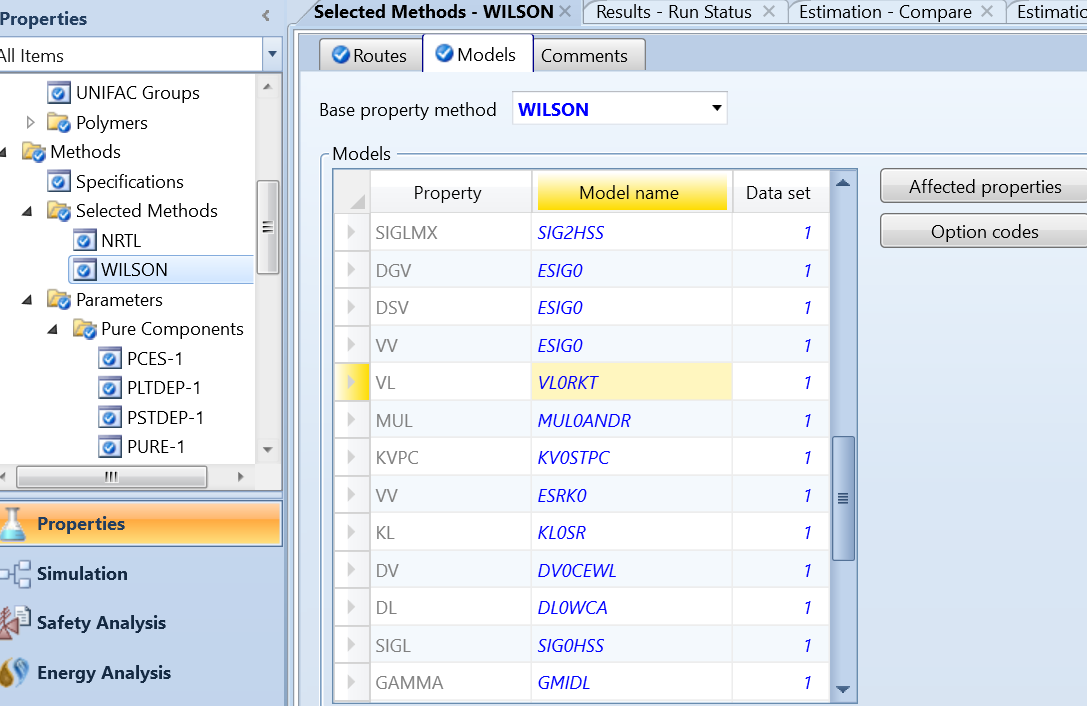 |
图6 修改wilson方程模型中预测密度(VL)的子模型VL0RKT

图7 修改wilson方程模型中预测密度(VL)子模型VL0RKT默认的Rackett公式为DIPPR公式

图8控制面板错误信息提示
表2 PPDS Campbell-Thodos方程参数表
|
Parameter Name/ Element |
Symbol |
Default |
MDS |
Lower Limit |
Upper Limit |
Units |
|---|---|---|---|---|---|---|
|
RACKET/1 |
C1 |
R*Tci/Pci |
— |
— |
— |
MOLE-VOLUME |
|
RACKET/2 |
C2 |
RKTZRA |
x |
0.1 |
1.0 |
— |
|
RACKET/3 |
C3 |
0 |
x |
0 |
0.11 |
— |
|
RACKET/4 |
Tmin |
0 |
x |
— |
— |
TEMPERATURE |
|
RACKET/5 |
Tmax |
1000 |
x |
— |
— |
TEMPERATURE |
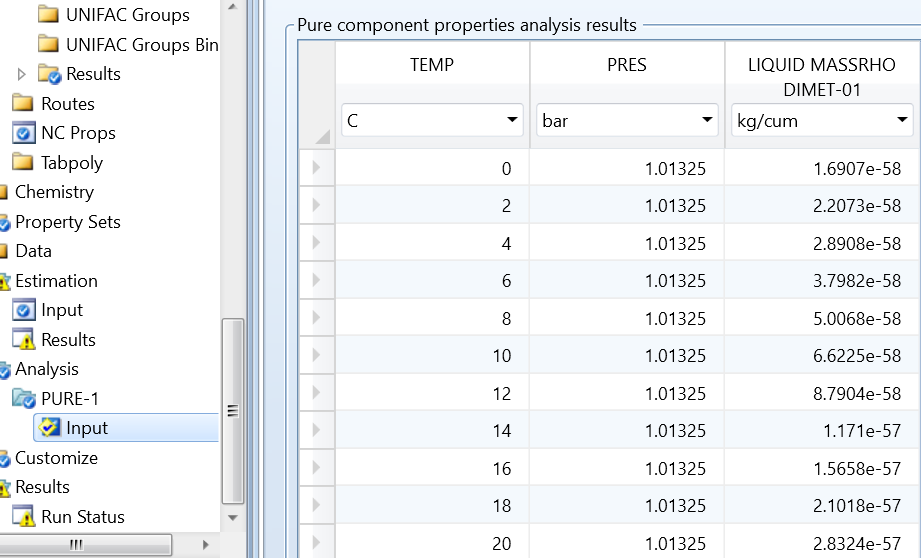
图9 由PPDS Compbell-Thodos估算的结果
由图8可知,结果显然更不靠谱。
至此,利用VL0RKT模型计算液体体积全部尝试完毕,结果全部不太满意。
我们尝试通过改变模型实现更满意的预测。在如图6中,VL0RKT有许多平行选项可选。如选择VL0CONS模型。
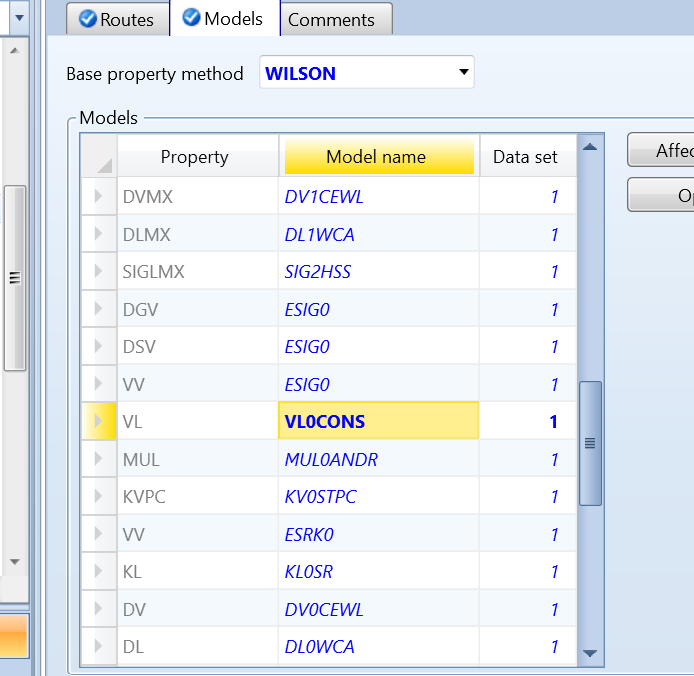
图10 在“Models”中将VL0RKT替代为VL0CONS
3 调整models中模型用VLCONS替代VL0RKT
操作如图10所示。该模型认为整个温度范围内,液体摩尔体积是一常数,这就要求在使用该模型之前,需要指定一个液体摩尔体积值VLCONS,如没有该值,系统会自动使用VL0RKT模型。如图11所示。在Methods>Parameters>Pure components>PURE-1中指定VLCONS值。
运行分析后,结果如图12所示。由图看出,在整个温度范围内,结果均为一常数1986。
图
 |
11 为VLCONS指定值
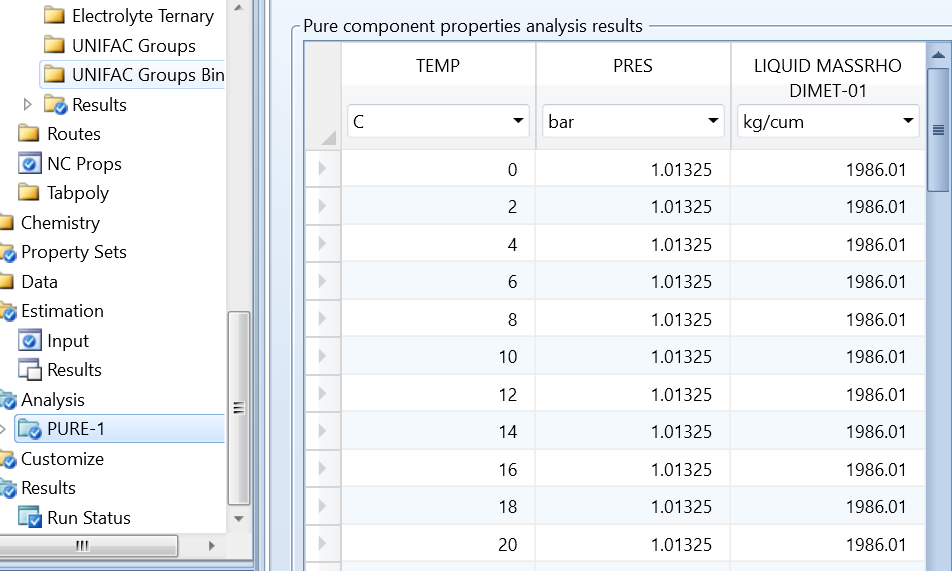
图12 运行VLCONS模型结果
4 调整models中模型用VL0MRK替代VL0RKT
按如3的操作步骤,将VL0RKT模型修改为VL0MRK,运行结果如图13所示。由图可知,结果显然是错误的。

图13 选用VL0MRK的运行结果
5 调整计算路径
上述都是在计算路径VL01下进行的,如得不到合适的结果,我们可以调整计算路径,将VL01改为其它形式,修改方法如图14所示。其默认计算公式如图15所示。选定以后运行程序,结果提示错误。
在控制面板中显示错误,如图16所示。
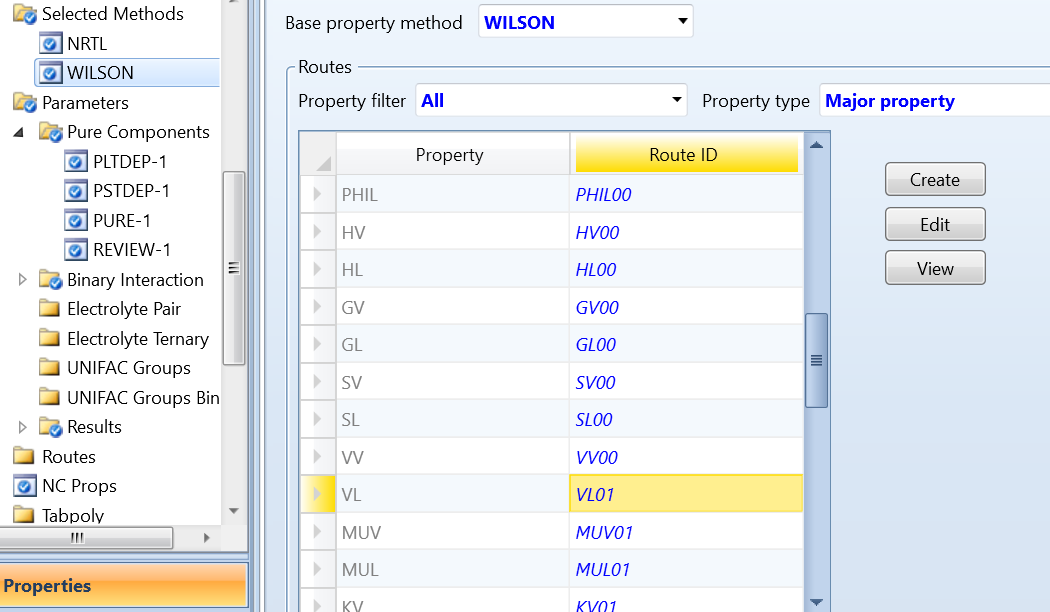
图14 修改计算路径
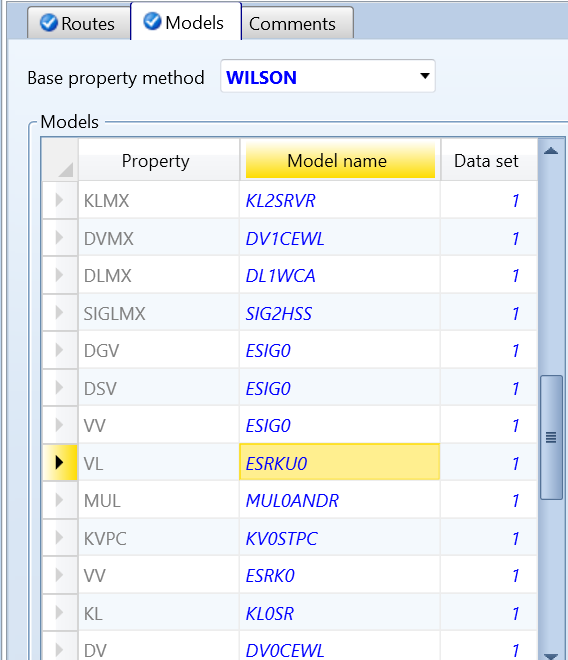
图15 VL20模型默认的计算公式

图16 控制面板的错误信息
改用其它路径,结果也是差强人意。
由以上看出,由于物性估算系统对物性的估算误差较大,导致所有方法预测液体密度对温度的变化误差均较大。
但由此例我们可以学习到调整模型中子模型方程的三种方法,即:1)在子模型中变更计算公式;2)变更子模型;3)变更子模型路径。
但三种方法的可靠性,均有赖于已知相关参数的准确性,因此,需要实验手段去提供输入数据,及后来的确认模型有效性。
需要说明的是,上述物性估算方法是在无法实验无现成文献情况下不得已采取的手段。对其结果要谨慎对待。如本例中,比较可靠的结果不是系统默认的模型计算结果,而是3中利用models中模型用VLCONS的计算结果比较可靠。
本例之所以误差较大,其原因在于,估算物性参数时,仅考虑基团的贡献,而未考虑cd离子对物性参数的贡献。
总之,在物性估算系统中,尽量采取实验数据进行物性参数的拟合。